First-principles calculations
of NMR parameters
First-principles calculations
of NMR parameters

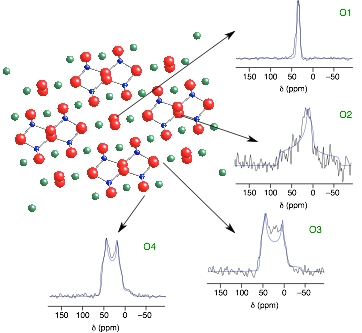
Calculated and experimental 17O NMR spectra of wadsleyite (β-Mg2SiO4)
Recently, much interest has focussed on the use of ab initio methods to predict from first principles the NMR chemical shifts and quadrupolar couplings in a given structure.
We use CASTEP (a planewave pseuodopotential code based on density functional theory (DFT)) to help assign our spectra, confirm parameters difficult to extract experimentally, interpret spectra and, in some cases, to predict spectra prior to acquisition.
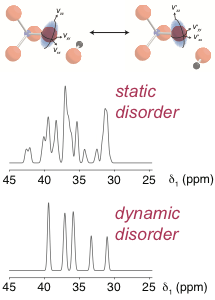
Calculated 17O NMR spectra of hydroxyl-clinohumite (4Mg2SiO4.Mg(OH)2)) assuming static or dynamic disorder of the hydroxyl groups
Current work includes the study of high-pressure silicate minerals, disorder in ceramic materials, such as pyrochlores, and the host-guest interactions in microporous frameworks.

19F NMR spectra of clinohumite (4Mg2SiO4.Mg(OH,F)2)) and assignment from DFT calculations
