Michael Bühl - Current
Research Interests
Chemical-Shift Computations for Transition-Metal
Nuclei
 With the development and implementation of suitable density-functional based
methods, NMR properties of transition-metal compounds can be calculated
with reasonable accuracy (review: M. Bühl, in: Calculation of NMR
and EPR Parameters. Theory and Applications, M. Kaupp et al. (Eds.),
Wiley-VCH, 2004; click on adjacent picture for more information ).
Current developments are focused on taking experimental conditions, that is,
temperature and solvent, explicitly into account in the computations.
With the development and implementation of suitable density-functional based
methods, NMR properties of transition-metal compounds can be calculated
with reasonable accuracy (review: M. Bühl, in: Calculation of NMR
and EPR Parameters. Theory and Applications, M. Kaupp et al. (Eds.),
Wiley-VCH, 2004; click on adjacent picture for more information ).
Current developments are focused on taking experimental conditions, that is,
temperature and solvent, explicitly into account in the computations.
Illustrative examples:
- Performance of Various Density Functionals:
In most cases, the B3LYP combination of density functionals is well suited
for the computation of transition metal NMR chemical shifts (M. Bühl,
Ann. Rep. NMR Spectrosc. 2008, 64, 77). In some cases, for
instance for 93Nb chemical shifts (M. Bühl, B. Wrackmeyer, Magn. Reson. Chem.
2010, 48, S61), non-hybrid
density functionals are preferable.
- Modelling Thermal and Medium Effects:
 A computational protocol to estimate thermal and solvent effects on transition
metal chemical shifts has been established (M. Bühl, M. Parrinello, Chem.
Eur. J., 2001, 7, 4487). The approach involves Car-Parrinello
molecular dynamics simulations of the substrate in a periodic water box
and chemical-shift calculations for a number
of snapshots along the trajectory. Substantial solvation effects are found, e.g.
for 59Co chemical shifts of ionic cobalt complexes such as
[Co(H2O)6]
3+ (M. Bühl et al., Chem. Eur. J. 2006, 12
, 477).
A computational protocol to estimate thermal and solvent effects on transition
metal chemical shifts has been established (M. Bühl, M. Parrinello, Chem.
Eur. J., 2001, 7, 4487). The approach involves Car-Parrinello
molecular dynamics simulations of the substrate in a periodic water box
and chemical-shift calculations for a number
of snapshots along the trajectory. Substantial solvation effects are found, e.g.
for 59Co chemical shifts of ionic cobalt complexes such as
[Co(H2O)6]
3+ (M. Bühl et al., Chem. Eur. J. 2006, 12
, 477).
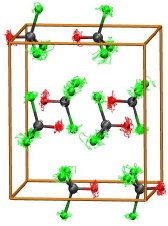
Such simulations have been extended to model the effects of a polar
crystal environment on chemical-shift and nuclear quadrupole-coupling tensors, e.g. for the
51V nucleus in solid VOCl3, see adjacent view of the unit cell
(R. Bjornsson et al, PCCP 2011, 13, 619). A recently proposed protocol for QM/MM optimizations of molecular crystals (R. Bjornsson et al, J. Chem. Theor. Comput. 2012, 8, 498) has been implemented in the latest release of the ChemShell software package.
- Transition-Metal NMR of Metalloenzymes:
|
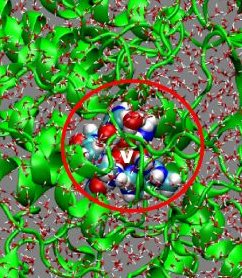
51V NMR tensor properties have been evaluated in a QM/MM framework
for vanadium-containg enzymes, namely vanadium-dependant halooperoxidases
(for a chloroperoxidase see the adjacent figure with a typical QM region highlighted as space-filling
model and the MM region, i.e. the remaining protein and surrounding solvent,
represented schematically). In combination with experimental solid-state NMR
data, elements of the chemical-shift and nuclear quadrupole-coupling
tensors derived by QM/MM computations for several models can afford insights
into structural details such as protonation state of the central vanadate
moiety and hydrogen-bond network in the active site (M. P. Waller et al,
Chem. Eur. J. 2007, 13, 4723; K. R. Geethalakshmi et al, J. Phys. Chem. B 2009
, 113, 4456).
|
Modeling of Homogeneous Catalysis
Rational design and optimization of catalysts requires detailed knowledge
of the reaction mechanism. The computational approach involves characterization
of the catalytic cycle by locating all relevant intermediates and
transition states. This information can help to identify the rate-determining step
and open up ways for a rational design of new, improved catalysts.
Illustrative examples:
- Alcohol dehydrogenation:
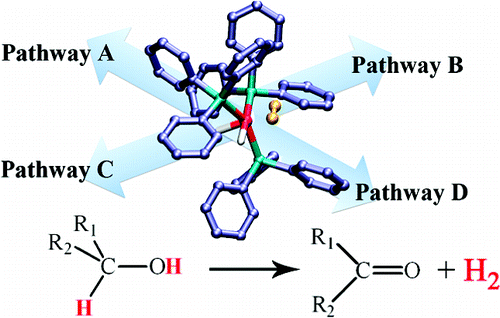
|
Alcohols are potential sources of hydrogen for the use in fuel cells.
Development of homogeneous catalysts for hydrogen release from alcohols
is a topical research area. A detailed computational study of a classic
Ru-based system reveals a multitude of competing, interlocked pathways
(see adjacent figure). The preferred pathway can depend on the substrate,
and the release of the latter from the metal center can be the rate-limiting
step (J. Am. Chem. Soc. 2010, 132, 8056).
Similarly detailed insights are obtained for alcohol decarbonylation, an
important competing side reaction (
N. Sieffert et al., Chem. Eur. J. 2014, 20, 4141).
|
- Peptide bond formation:
|
The high mechanical and thermal stability of bacterial surface proteins
is related to an emerging building block in biochemistry, namely the
isopeptide bond, an additional amide bond cross-linking the regular peptide
backbone. The catalytic role of a nearby residue in the formation of this
bond has been rationalized through QM/MM calculations (R. M. Hagan et al., Angew. Chem. Int. Ed.
2010, 49 , 8421).
|

|
Molecular Dynamics of Transition
Metal Complexes
First-principles MD simulations are
a valuable tool to study the dynamics of chemical systems on the picosecond
time scale. Rapid and spontaneous rearrangement processes in fluxional molecules
can be directly followed and analyzed. Slower processes can be studied using
constrained MD simulations along a predefined reaction coordinate, which allows
extraction of free energies via thermodynamic integration.
Illustrative examples:
- Protonated Ferrocene:
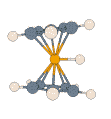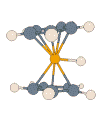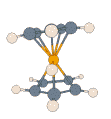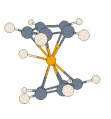
|
For this prototypical product of electrophilic attack on a metal complex,
details of the potential energy surface and, thus, the dynamic processes on
it, can depend noticeably on the particular density functional and basis set
employed. All methods agree that the extra proton is highly fluxional, and
that there is rapid interconversion between structures described as metal-protonated
and those with an agostic interaction (M. Bühl, S. Grigoleit, Organometallics
2005, 24, 1516). In the adjacent animation one can see how,
during 2 ps of MD at the BP86/SVP level, the proton bends from the initial
metal-protonation site toward a ring, and is reversibly transferred between
rings while moving around the perimeter.
|
-
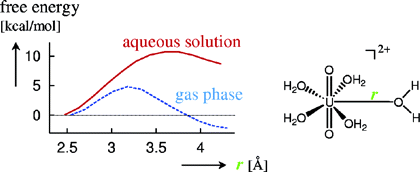
Uranyl Complexes:
Structure, speciation, and ligand binding energies of uranyl(VI) hydrate
in aqueous solution are studied with
constrained CPMD simulations and thermodynamic integration.
According to the free-energy profiles shown below, four-coordination is to be expected
for this complex in the gas phase, but there is a strong reinforcement of the U-O bonds in
water, where the five-coordinated form is clearly preferred (M. Bühl
et al, J. Am. Chem. Soc. 2005, 127, 13506). This reinforcement
of the U-water bond upon solvation can be rationalized through cooperative polarization
effects (M. Bühl et al, Inorg. Chem. 2011, 50, 299).
Similar simulations have been used to reproduce and predict binding constants of various anionic ligands to uranyl in water, and have helped to reconcile seemingly conflicting experimental results on an intriguing exchange process in aqueous uranyl hydroxide (see a review on MD simulations of uranyl complexes: M. Bühl and G. Wipff, ChemPhysChem 2011, 12, 3095. These simulations have also been extend to nonaqueous solvents, including liquid ammonia (P. Woidy, M. Bühl, F. Kraus, Dalton Trans. 2015, 44, 7332).
List of publications
Last update: 11-Feb-2016/mb